CicArVarDB contains the pages listed and described as follows:
Home: Introductory page for the database.
Search: Main search page which facilitates in mining the variation data stored in the database.
BLAST: The simplest way to identify if the sequence of interest already exists as a variation.
JBrowse: Helps in the visualization of the chickpea genome with the feature like gene, CDS, non coding regions and the variations found across each genotype.
Tutorial: The page detailing on “How to use this database!”.
Resources: This page provides the links to other genomic/transcriptomic repositories relevant to the crop chickpea.
Contact us: The user may contact us in case of any queries/problems.
Home: Introductory page to the database. The home page provides a brief introduction about the database and the contents present in it. It archives over a 1.9 million variations that includes SNPs, small Insertion and Deletion (1-10 basepair) spanning across the pseudomolecules of the chickpea genome, discovered by aligning 29 whole genome re-sequenced (WGRS) and 61 restriction site-associated DNA sequenced (RAD) lines of chickpea to the CDC-Frontier reference genome. The SNPs were reported in case it was present across any two genotypes and SNP quality greater than 25. The home page contains links to other pages like ‘search page’, ‘BLAST’, ‘JBrowse’, etc, in the database.
Search: The search page provides an easy-to-use interface to the user to extract variations stored in the database. It lists two functionalities, basic and advanced search option for the user to retrieve the data of their interest. The user can switch to either of two search modes by selecting respective options.
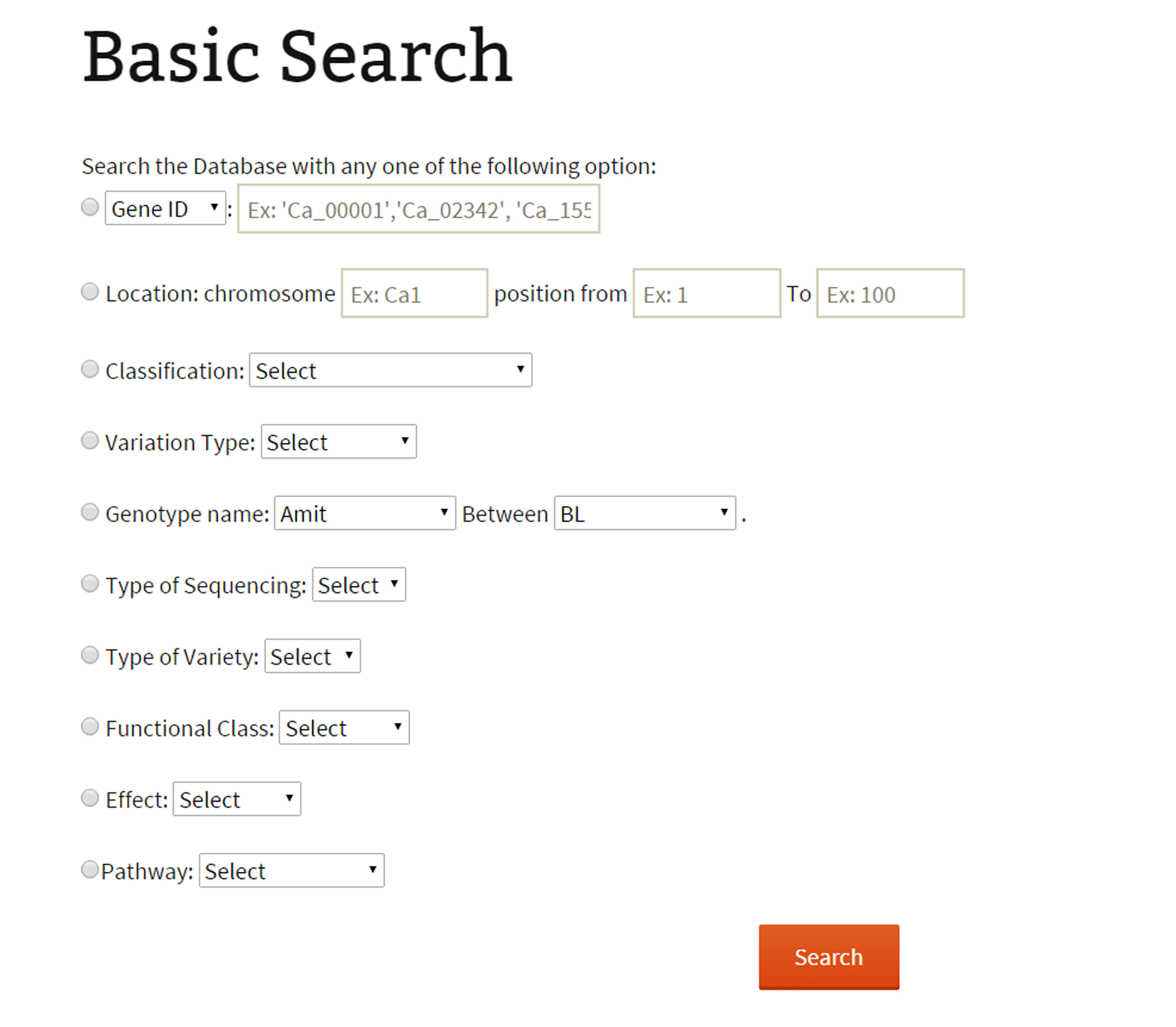
Basic search:The basic search provides options to narrow down the reported variations. The user has different options to perform a variations search. The main objective of basic search has been to let the user retrieve information based on just one criteria. The user, if limited to just one information like GeneID, SNP-ID as explained below:
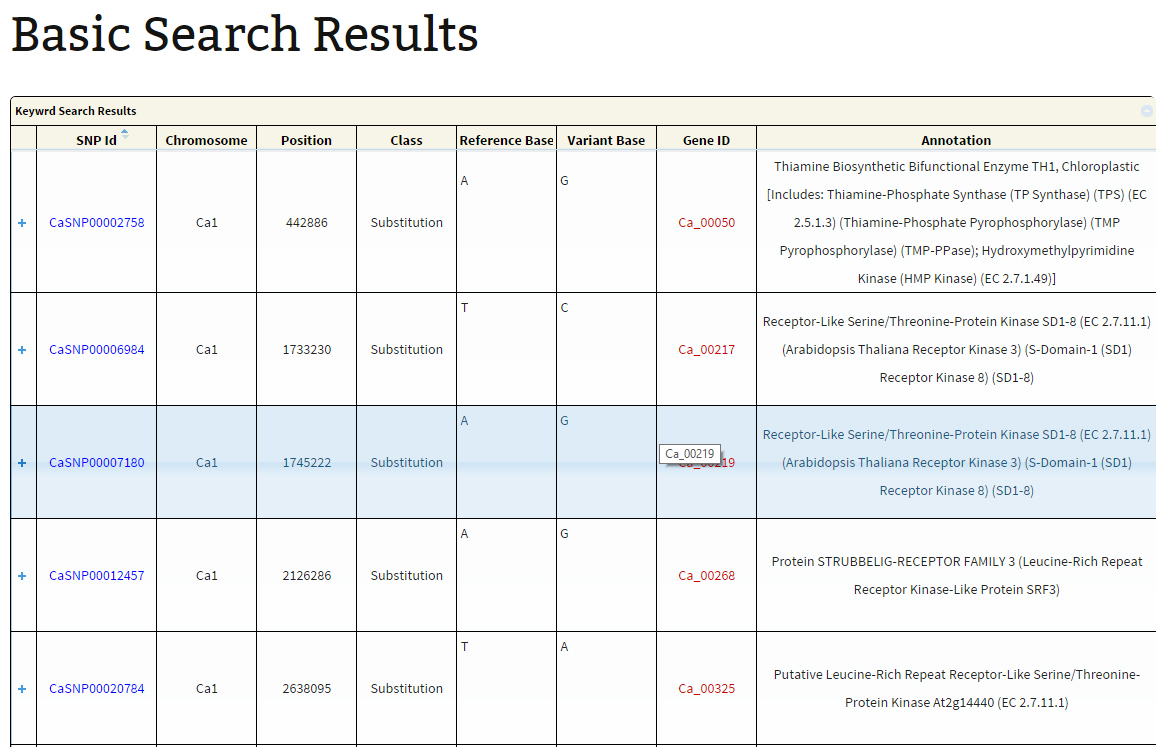
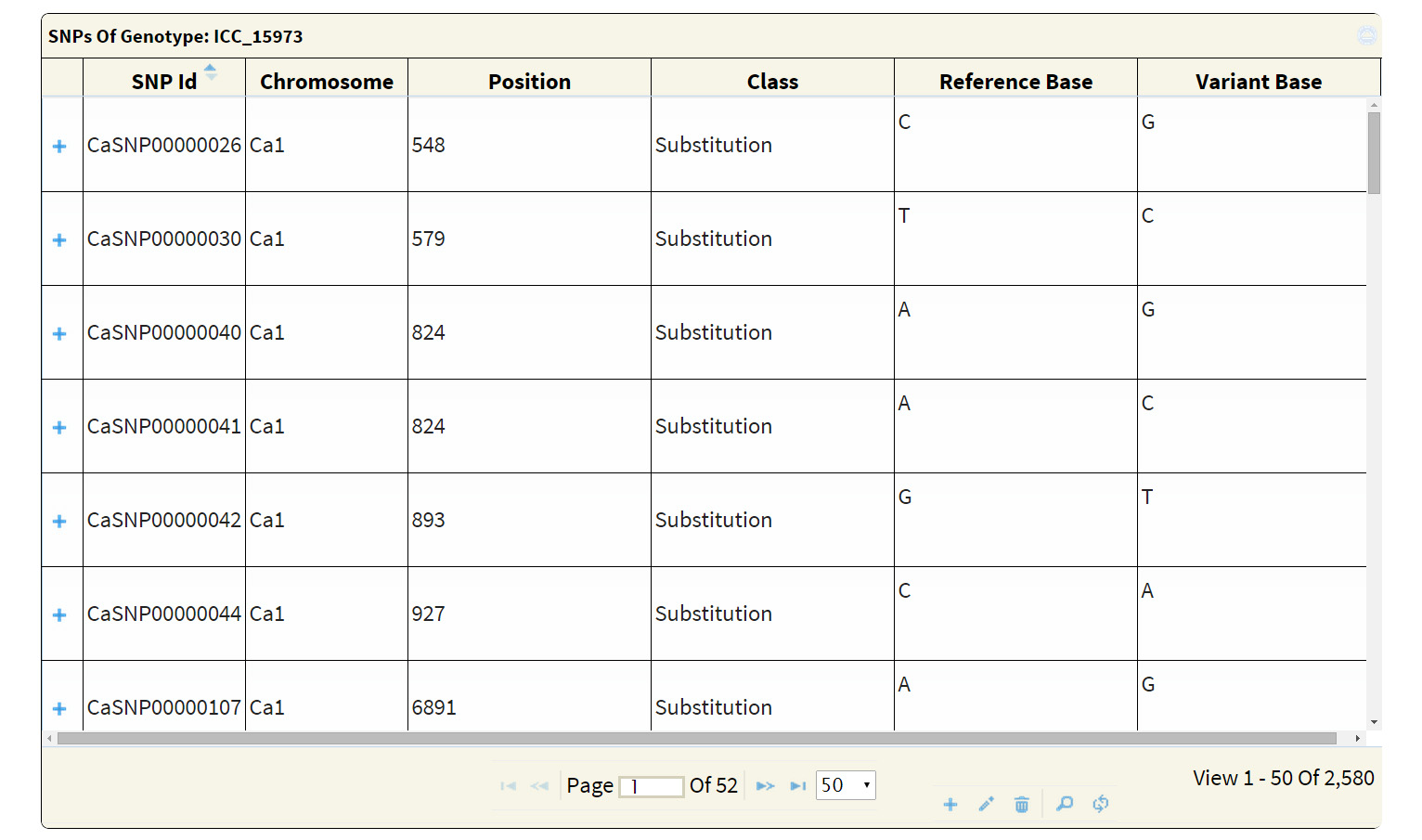
The search output reports the location of variations along with the geneID if the variation is present within the coordinates of a gene. A click on ‘+’ symbol present at the beginning of each record expands the row with 2 columns displaying genotype names which match to the reference and variant base respectively. The hyperlink on the respective SNP-ID leads to a JBrowse page for the visualization of variation and the respective GeneID leads to the annotation page which links to Uniprot details, gene ontology information and KEGG ontology, etc.
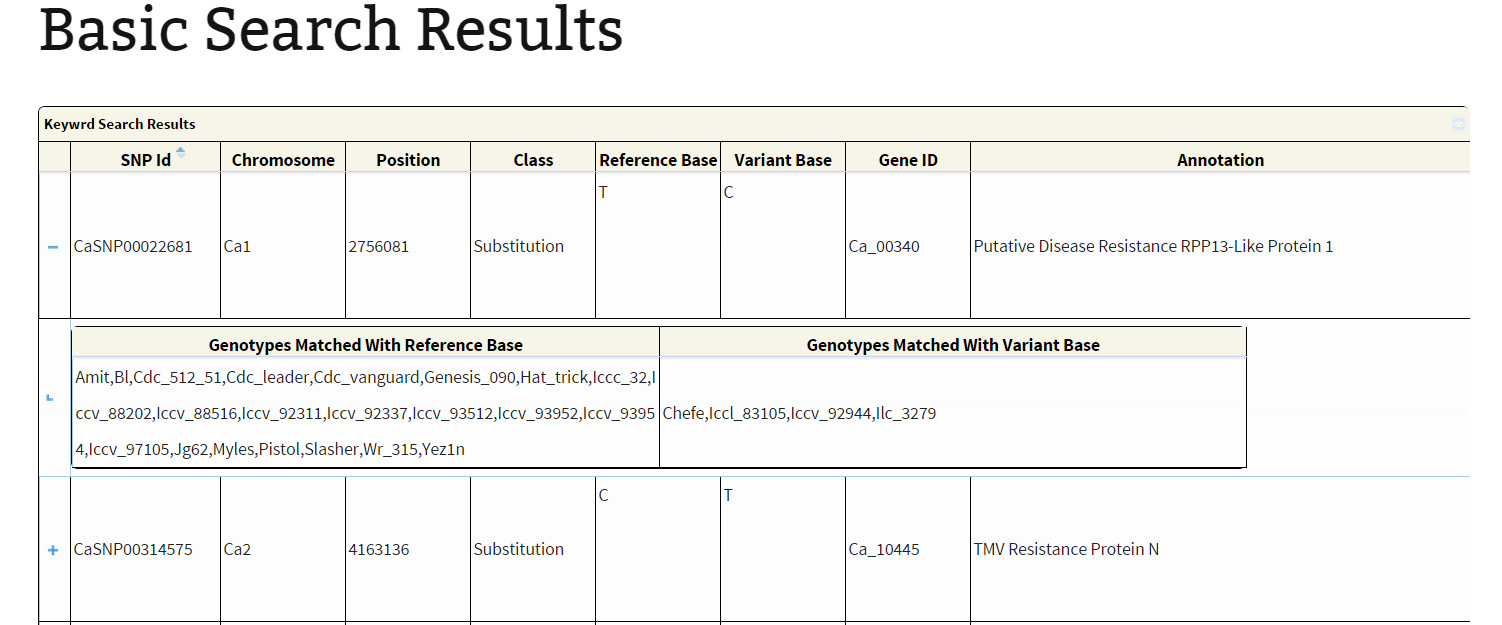 Export option is provided to export the results
Export option is provided to export the results
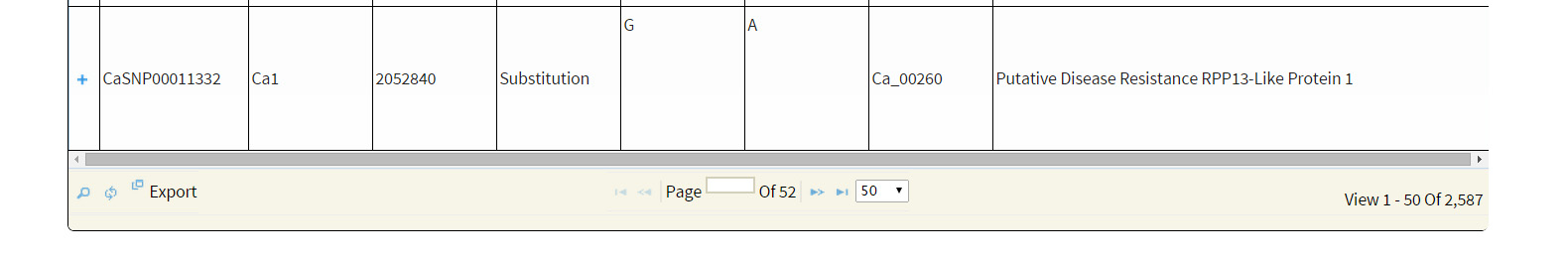
Keyword search: Variations can be searched based on the
GeneID: We have retained the same GeneID as in the generic feature file (gff) provided with Varshney et al 2013. GeneID begins with the string ‘Ca_’ followed by a five digit number from ‘00001’ to ‘28269’. For example ‘Ca_00001′, ‘Ca_02322’, ‘Ca_25269’ are the GeneID the user can make use of. Search is case insensitive, so the user can even search by the string ‘CA_’, which will yield the same search results.
SNP-ID: Variations are identified by the respective IDs. IDs starts with string ‘CaSNP’ followed by a seven digit number from 0000001 to 01965803. Example: SNP Ids are ‘CaSNP00000001’, ‘CaSNP00000003’, CaSNP00010003’, CaSNP00200003’, ‘CaSNP01965803’ etc. Here, again the search is case insensitive.
Keyword: The keywords such as ‘disease resistance’, ‘kinase’ ‘transporter’, TMV, Leucine-rich, etc. Variation search using keywords is supported by boolean operators such as ‘AND’, ‘OR’, ’NOT’ for a combination of words, with space as delimiter, supporting search for the exact string match. For example: The user may search with terms ‘transporter’ ‘AND’ ‘sugar’ which yields variations affecting genes annotated with terms transporter and sugar. To search variations contained in genes annotated with terms tranporter but not containing sugar, the user need to search like ‘Transporter’ ‘AND NOT’ ‘Sugar’. The keyword search also supports search for a substring of a given single keyword (no spaces). For example: single word like ‘Kin’ can be used to search for ‘Kin11’, ‘Kinase’ etc.
 Location: Users can search for the presence of variations within specified genome co-ordinates. Chickpea draft genome from Varshney et al 2013 has 8 pseudo- molecules and name start with the string Ca and followed by single digit number 1 to 8 example Ca1, Ca2, Ca3, etc. User can enter the start and end position of the genome region of interest to fetch the SNPs. For example: to search variations between 1Mb regions to 5 Mb of pseudomolecule Ca8, the user need to enter start coordinate as 1000000 and end coordinate as 5000000 along with entry of Ca8 in the ‘Chromosome’ box.
Location: Users can search for the presence of variations within specified genome co-ordinates. Chickpea draft genome from Varshney et al 2013 has 8 pseudo- molecules and name start with the string Ca and followed by single digit number 1 to 8 example Ca1, Ca2, Ca3, etc. User can enter the start and end position of the genome region of interest to fetch the SNPs. For example: to search variations between 1Mb regions to 5 Mb of pseudomolecule Ca8, the user need to enter start coordinate as 1000000 and end coordinate as 5000000 along with entry of Ca8 in the ‘Chromosome’ box.
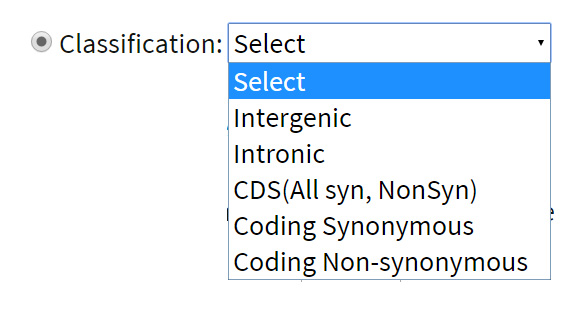
 SNP classification:Variations are classified according to their location in the genome such as intergenic, intronic and CDS. Variations within the CDS regions are further classified into synonymous and non-synonymous categories. Users can search for variations within these categories. Example: if the user wish to search for all non synonymous variations across the genome he just needs to go for the option ‘Coding Non Synonymous’.
SNP classification:Variations are classified according to their location in the genome such as intergenic, intronic and CDS. Variations within the CDS regions are further classified into synonymous and non-synonymous categories. Users can search for variations within these categories. Example: if the user wish to search for all non synonymous variations across the genome he just needs to go for the option ‘Coding Non Synonymous’.
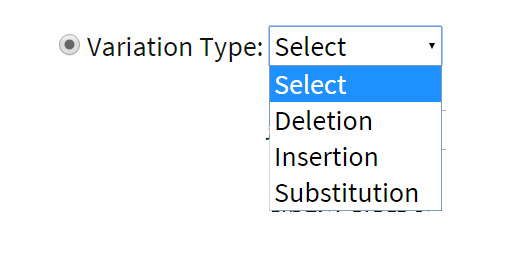
 Type of variants: Variations are categorized as insertions, deletions and substitutions to facilitate the search of variants of choice. For example: The user may only wish to search for the deletions present across the genome and can retrieve them by seletion of ‘deletions’ option.
Type of variants: Variations are categorized as insertions, deletions and substitutions to facilitate the search of variants of choice. For example: The user may only wish to search for the deletions present across the genome and can retrieve them by seletion of ‘deletions’ option.
 Between any two genotypes: Search can be executed for variations between any two genotypes of interest. This actually is a pairwise comparison of two genotypes having variation at the same position in the genome. The user can select two genotypes from the ones listed. Example: The user can get all variations between ‘Amit’ and ‘BL’ by selecting both of them one by one.
Between any two genotypes: Search can be executed for variations between any two genotypes of interest. This actually is a pairwise comparison of two genotypes having variation at the same position in the genome. The user can select two genotypes from the ones listed. Example: The user can get all variations between ‘Amit’ and ‘BL’ by selecting both of them one by one.
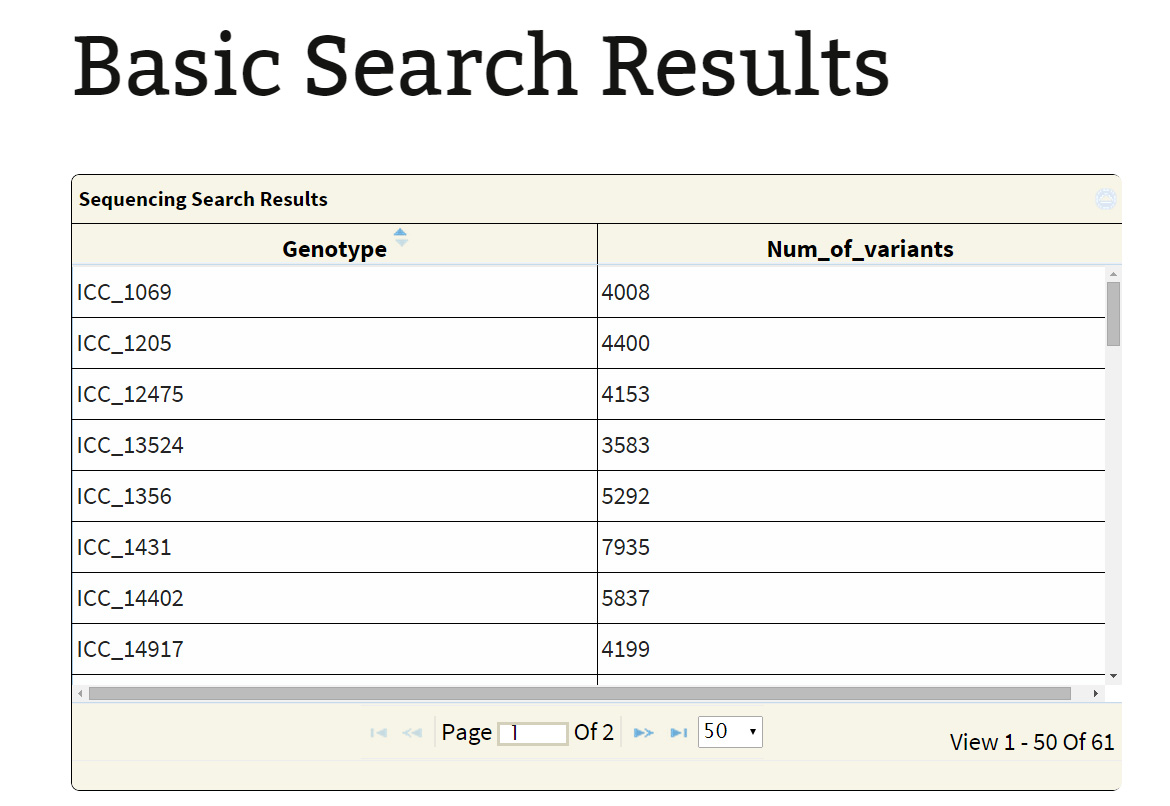
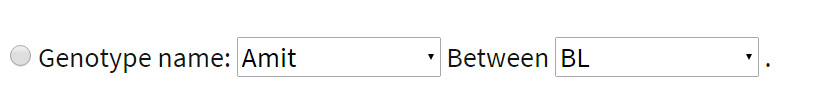 Sequencing type: Currently, the database hosts variations obtained from WGRS and RAD sequencing data. If the user is only interested in variations in the RAD sequenced lines he may get them by selecting the option “RAD”. Selection of the type of sequencing either WGRS or RAD leads to a table containing the genotypes and number of variants found. Selection of particular genotypes leads to retrieval of variations information on the sub table.
Sequencing type: Currently, the database hosts variations obtained from WGRS and RAD sequencing data. If the user is only interested in variations in the RAD sequenced lines he may get them by selecting the option “RAD”. Selection of the type of sequencing either WGRS or RAD leads to a table containing the genotypes and number of variants found. Selection of particular genotypes leads to retrieval of variations information on the sub table.

 Type of genotypes: Sequenced desi and kabuli chickpea genotypes can be filtered for variants occurring in either of these types. For example: The user may filter the variations occurring in only desi genotypes when compared to the CDC-frontier reference genome which is a Kabuli type variety. Selection of type of variety leads to a table containing the genotype name and the number of variations found in it. The variation information for the particular type of genotype can be retrieved by clicking on the genotype.
Type of genotypes: Sequenced desi and kabuli chickpea genotypes can be filtered for variants occurring in either of these types. For example: The user may filter the variations occurring in only desi genotypes when compared to the CDC-frontier reference genome which is a Kabuli type variety. Selection of type of variety leads to a table containing the genotype name and the number of variations found in it. The variation information for the particular type of genotype can be retrieved by clicking on the genotype.
 Functional class: SNPs classified as missense, nonsense and silent can be retrieved. The users are particularly interested in the variations having non-sense or mis-sense effect due the change. This may directly affect a trait a genotype may be linked to.
Functional class: SNPs classified as missense, nonsense and silent can be retrieved. The users are particularly interested in the variations having non-sense or mis-sense effect due the change. This may directly affect a trait a genotype may be linked to.
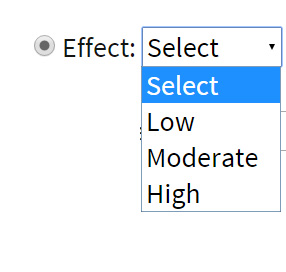
 Effect intensity: On the basis of the severity of their effects, variations were differentiated into low, moderate and high effect variations. These variations may occur in the intergenic, intronic or exonic regions in the genome. High effect variations may be targeted for the improvement of the varieties.
Effect intensity: On the basis of the severity of their effects, variations were differentiated into low, moderate and high effect variations. These variations may occur in the intergenic, intronic or exonic regions in the genome. High effect variations may be targeted for the improvement of the varieties.
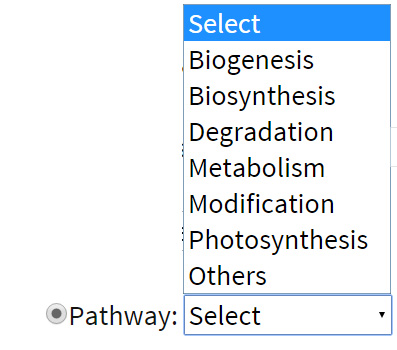
 Pathway: Search can be carried out for the variations affecting a gene featured in a specific biological pathway. For example: User may retrieve the variables affecting the metabolism pathway in the system.
Pathway: Search can be carried out for the variations affecting a gene featured in a specific biological pathway. For example: User may retrieve the variables affecting the metabolism pathway in the system.
 Advanced search:
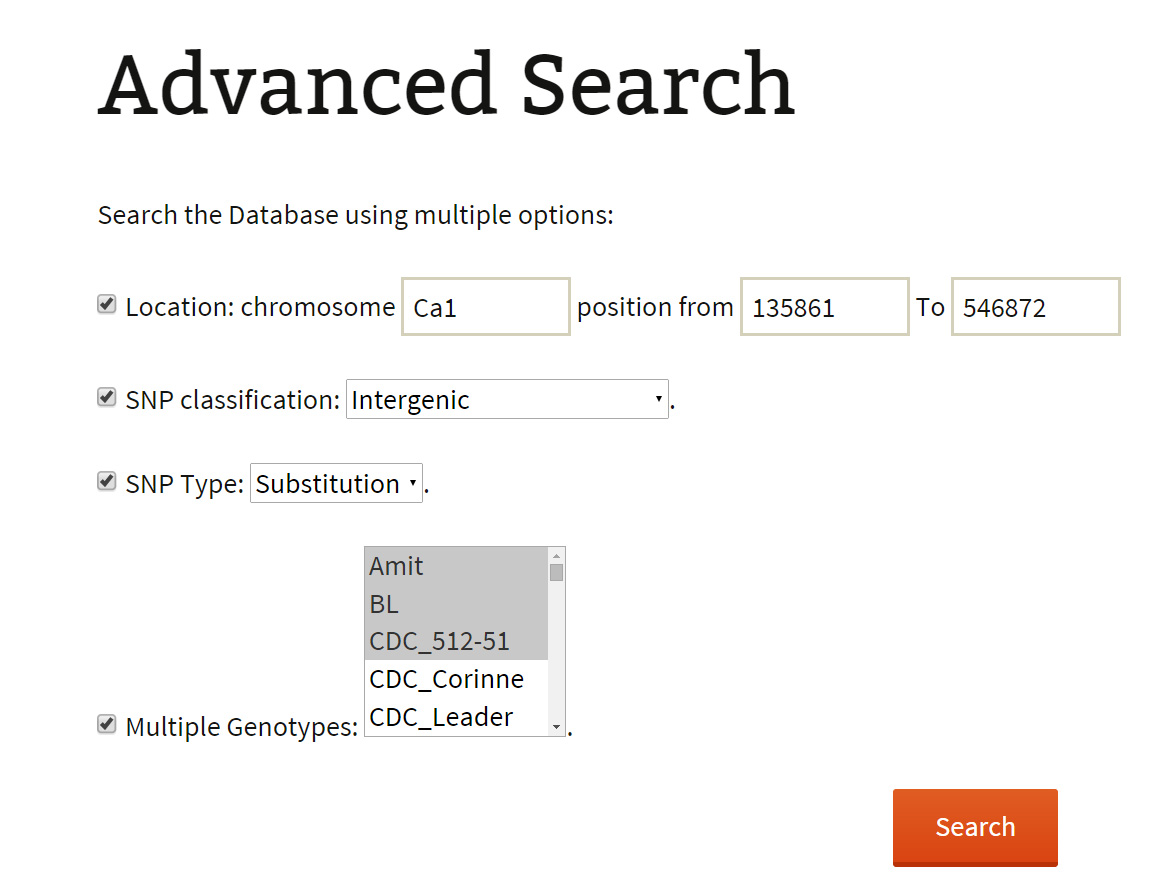
An advanced search method provides the user with more comprehensive selection criteria to choose at least any two of the listed options:
Advanced search:
An advanced search method provides the user with more comprehensive selection criteria to choose at least any two of the listed options:
An advanced search can be used to attain very specific and precise number of variations. The results of both basic and advanced searches includes a table of genomic co-ordinates, bases present at the SNP position in reference genome sequence and corresponding variant base call. A click on ‘+’ symbol present at the beginning of each record expands the row with 2 columns displaying genotype names which match to the reference and the variant base respectively. Variation can be visualized in JBrowse by clicking on the SNP ID provided in the results table. The basic search method numbers 6 and 7 list out the genotypes depending on the sequencing type (WGRS/RAD) and the type (desi/kabuli) of genotype respectively. Selecting a particular genotype from this table produces a table detailing the variations present in it.
For example: The user has the flexibility to combine 2 and more options listed in the search criteria. The user may extract for the genic variations across the pseudo-molecule ‘Ca1’. Or, he may wish to get all the deletions in a single pseudo-molecule. Similarly, he can mine for the variations (SNPs/InDels) present on pseudo-molecule Ca1 in two kabuli/desi varieties of his interest.
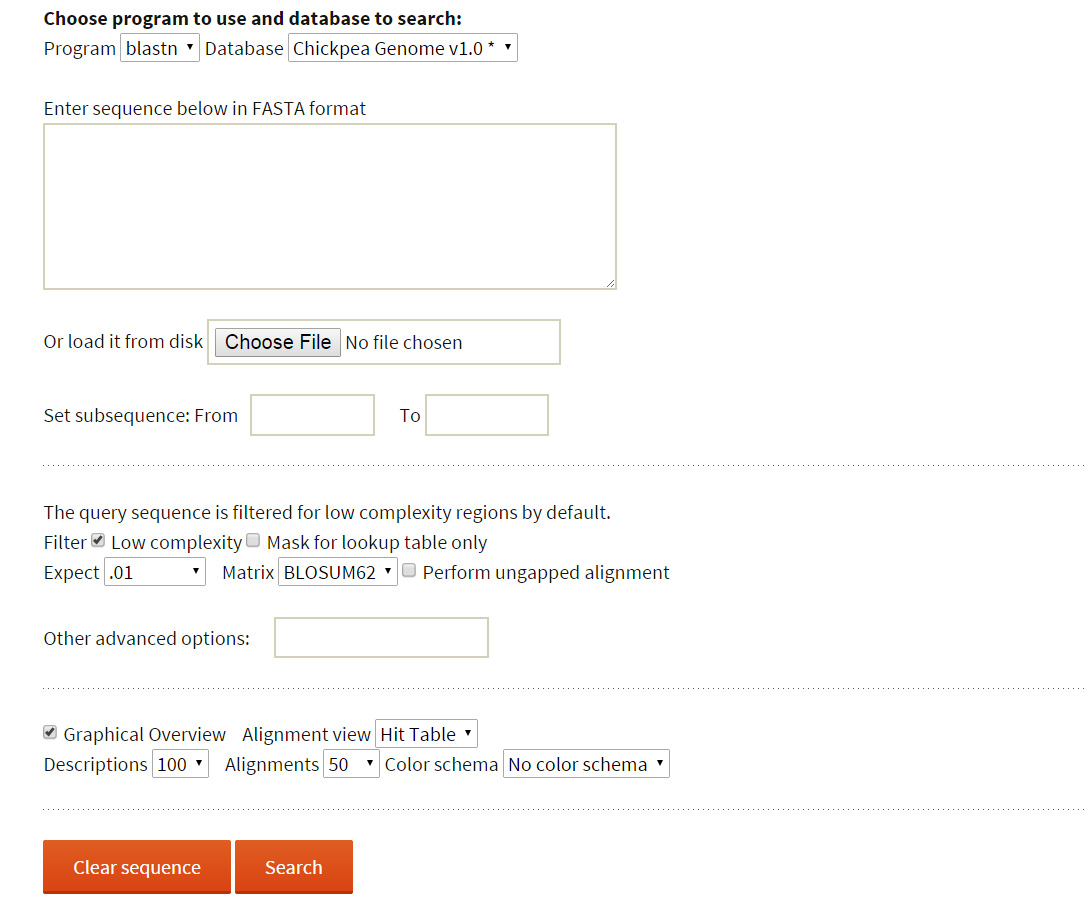
 BLAST: The user may wish to know the presence of reported variations in a sequence of interest. To perform this, webBLAST has been implemented as an additional tool to help the user find a homologous region in the reference sequence with a submitted query sequence and explore the presence of variations within a 10kb flanking region.
BLAST: The user may wish to know the presence of reported variations in a sequence of interest. To perform this, webBLAST has been implemented as an additional tool to help the user find a homologous region in the reference sequence with a submitted query sequence and explore the presence of variations within a 10kb flanking region.
 JBrowse:
JBrowse, a Java based genome browser is embedded in the database. JBrowser uses a client side scripting which makes the browser faster and allows easy scaling of large genome regions. JBrowse enables the users to upload BAM files as tracks to visualize alignment files faster as compared to the CGI based genome browsers and provides pictorial representation for the presence of variations.
JBrowse:
JBrowse, a Java based genome browser is embedded in the database. JBrowser uses a client side scripting which makes the browser faster and allows easy scaling of large genome regions. JBrowse enables the users to upload BAM files as tracks to visualize alignment files faster as compared to the CGI based genome browsers and provides pictorial representation for the presence of variations.
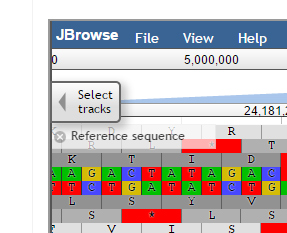
JBrowse has been customized for the ease of end user. We have added a ‘Select Tracks’ button which displays a popup page ‘Select Tracks’ where user can select multiple genotypes from the 90 genotypes listed along with their names, sequencing type and type of variety.
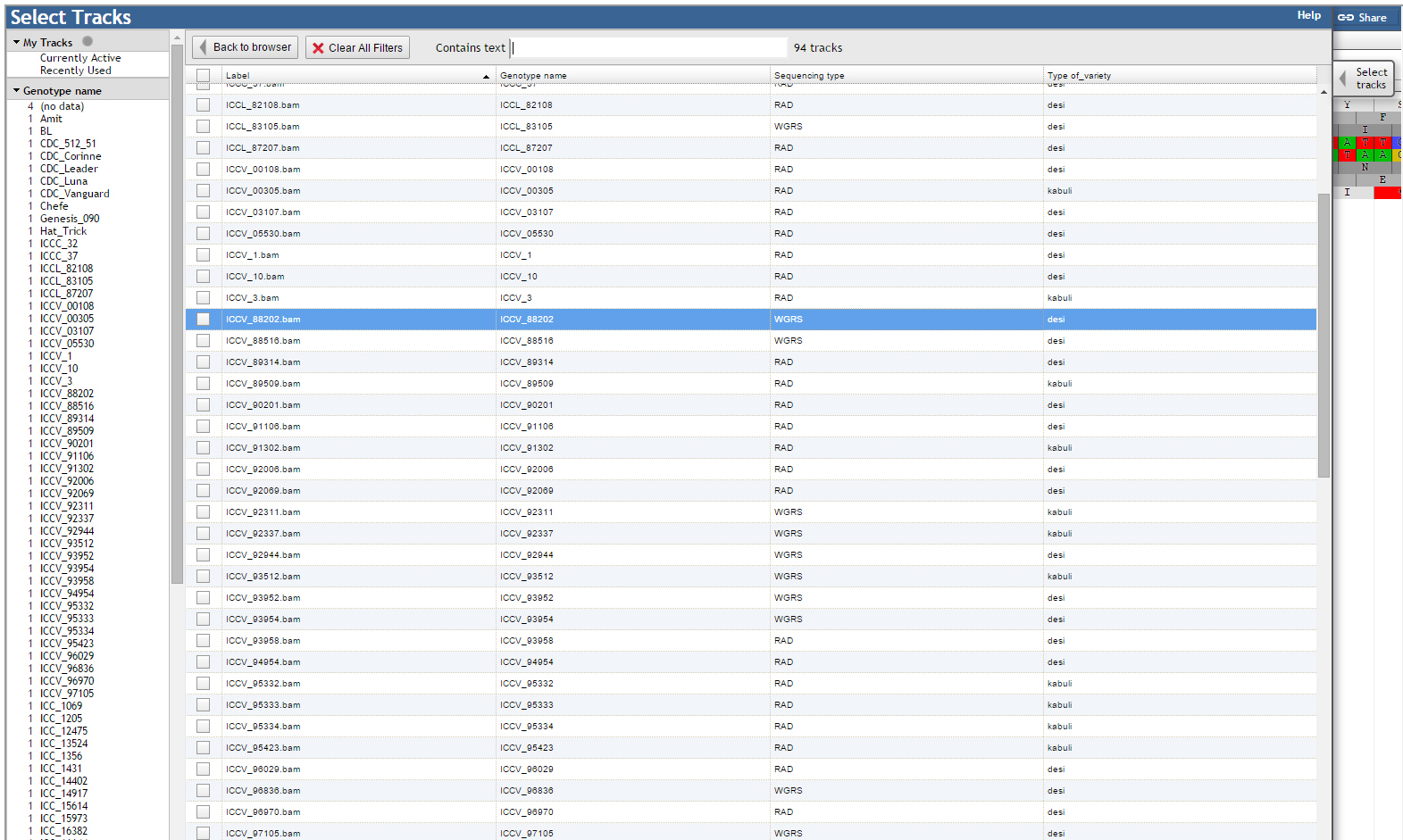
User may select all the genotypes as tracks, though by default JBrowse supports a maximum of 30 tracks. Tabs provided on the display popup page has the option of sorting.
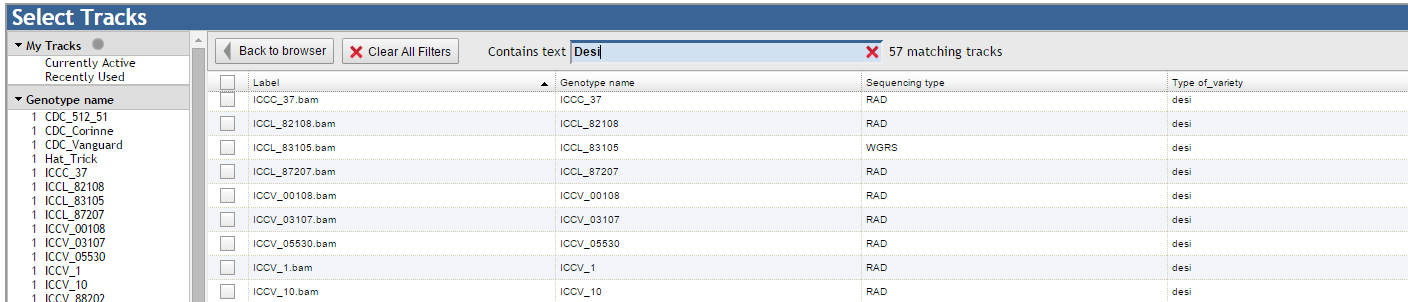
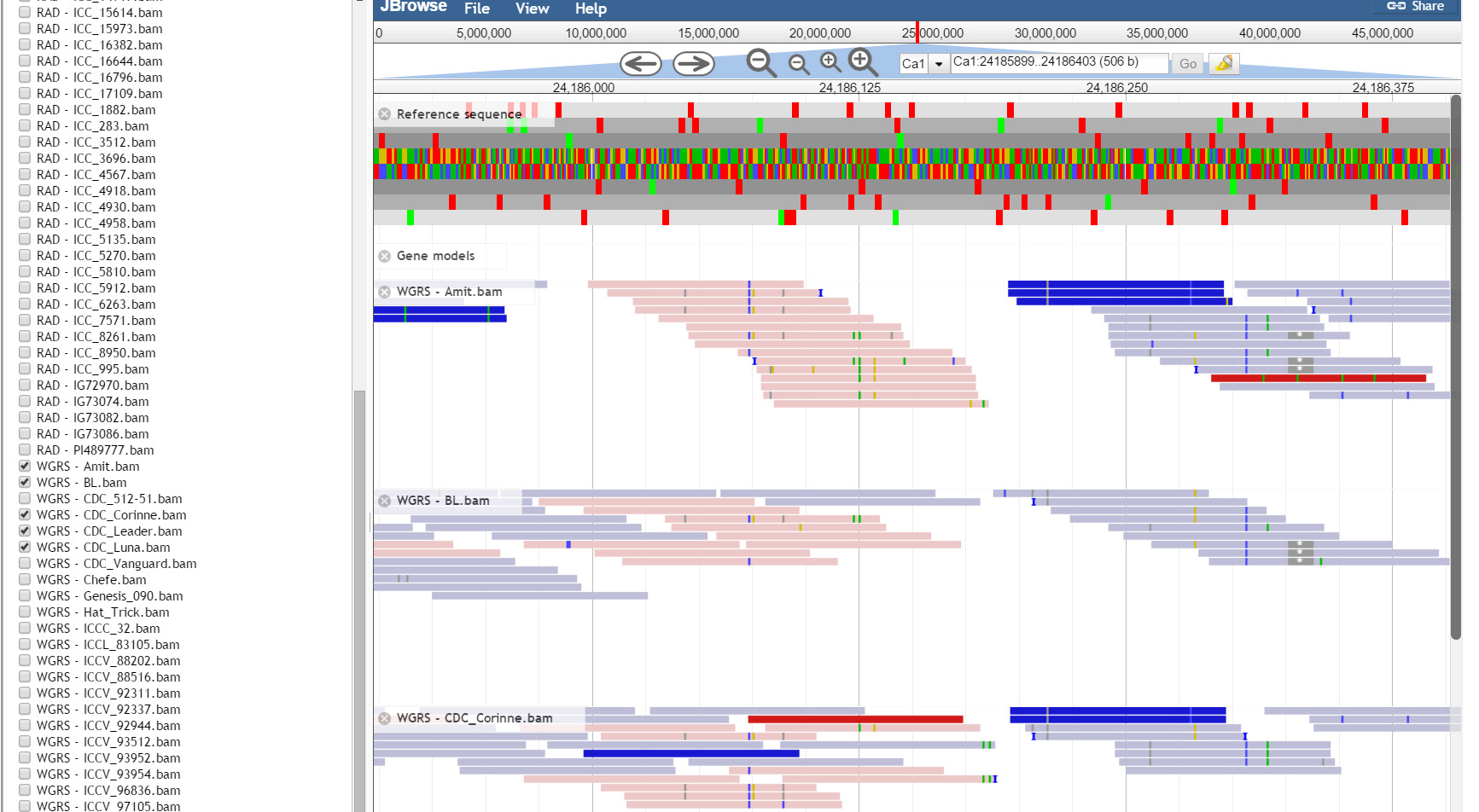
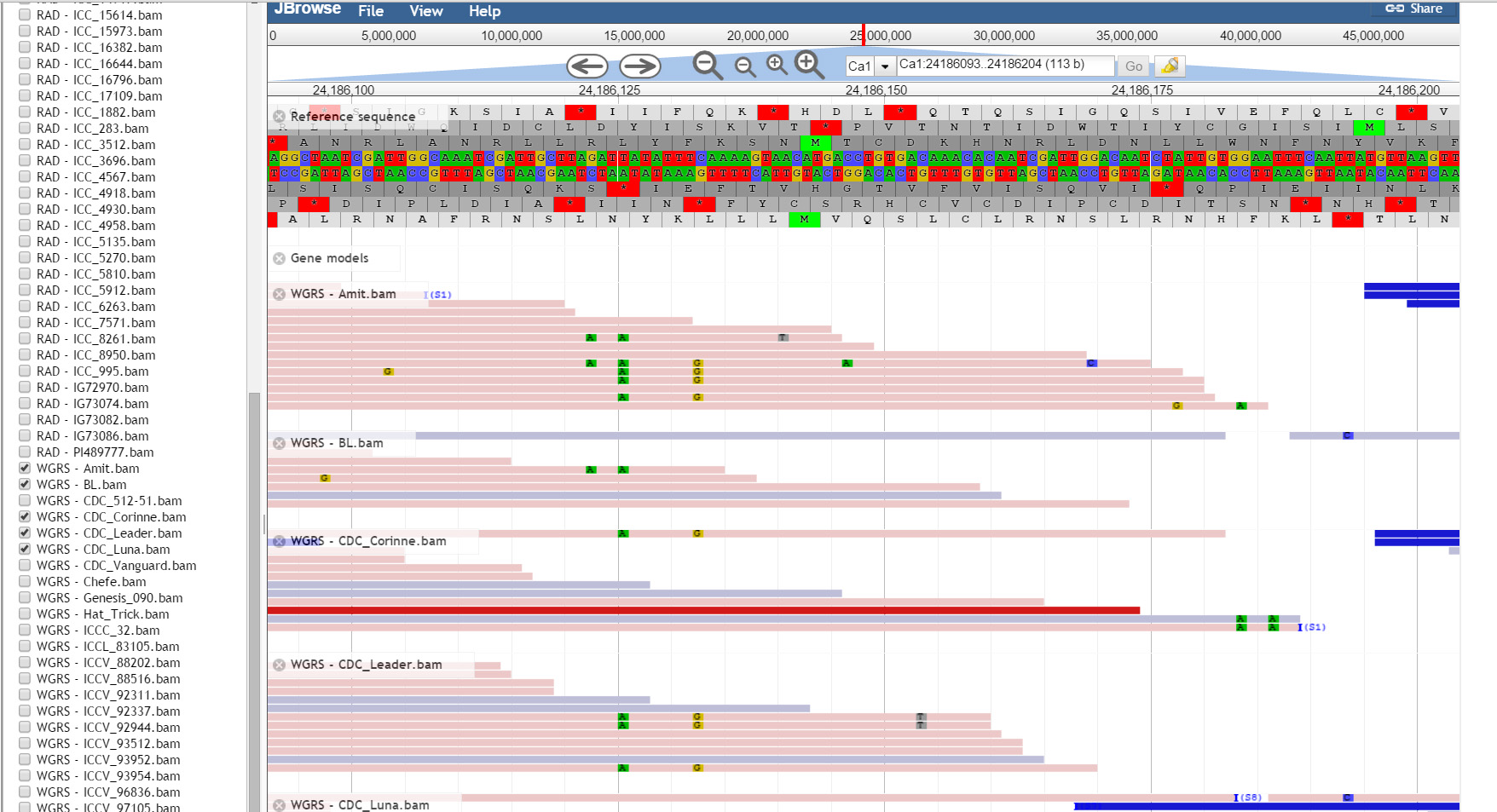
Resources:
This page provides the links to other genomic/transcriptomic repositories relevant to the crop chickpea.
Tutorial: The page detailing on “How to use this database!”.
Contact us:
